 Back Back
Research Article
ScienceAsia 28 (2002) :167-172 |doi: 10.2306/scienceasia1513-1874.2002.28.167
Ab Initio Molecular Orbital Computation Studies
of Ag+-C2H4 Complexation in the Presence of Water
Anawat Sungpet
ABSTRACT: Ab initio calculations are done to explore the influence of water on the Ag+-C2H4 complex formation. The simulated coordination environment of Ag ion is based on the existence of a dynamic equilibrium between the coordination of Ag ion with water and a sulfonate group, the counter ion. Calculated electronic properties reveal that electron delocalization from water molecules and the sulfonate group onto Ag 5s-atomic orbital reduces the ability of Ag ion to accept additional electrons from the C2H4 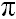 orbital. The dissociation of water molecules from the hydrated Ag+ is essential for the thermodynamic possibility of the Ag-C2H4 complex formation. It is also evident from the electronic structure calculations that the dissociation of water is favorable for the formation of a stable Ag-C2H4 complex. In the absence of water, the reaction between Ag ion strongly bound to sulfonate group and C2H4 is thermodynamically impossible. orbital. The dissociation of water molecules from the hydrated Ag+ is essential for the thermodynamic possibility of the Ag-C2H4 complex formation. It is also evident from the electronic structure calculations that the dissociation of water is favorable for the formation of a stable Ag-C2H4 complex. In the absence of water, the reaction between Ag ion strongly bound to sulfonate group and C2H4 is thermodynamically impossible.
Download PDF
Department of Chemical Engineering, King Mongkut’s University of Technology Thonburi, Bangkok 10140, Thailand.
* Corresponding author, E-mail: anawat.sun@kmutt.ac.th
Received 9 May 2001,
Accepted 15 Oct 2001
| 
